Fast facts
- Annotating genomic variants is a complex process
- Commonly used tools in this space have accuracy and/or scaling limitations
- Newly added to Emedgene, and available in DRAGEN Secondary Analysis, Illumina Connected Annotations provides exceptional performance in this field
- Top population studies, including All of Us and UK Biobank, rely on this tool
Background
The capability to reliably detect the millions of genetic variants present in every person has driven an incredible surge in the use of whole-genome sequencing. To begin making sense of all these variants, they must be annotated with useful information, like gene models, population frequencies, impact predictions, and reference databases. From hereditary genetic screening to oncology and population genetic studies, being able to annotate processed data accurately and quickly is key to any successful genomics project.
As two of the most ambitious population genomics programs ever conceived, the All of Us Research Program and the UK Biobank project both faced annotation challenges of epic proportions. Given hundreds of thousands of individuals and over a billion unique genomic variants to annotate, they needed software that met exceptionally high institutional standards of efficiency and accuracy. After evaluating multiple options, both groups selected Illumina’s annotator as their tool of choice because of its unique ability to fulfill their needs.1,2 For a recent release, UK Biobank successfully annotated their entire dataset of 500,000 whole-genome multi-sample variant call files in approximately 90 minutes.3
To empower scientists using Illumina sequencers, our bioinformatics team has been building software to address annotation needs for years, and we are proud to release our latest version, Illumina Connected Annotations. Built on the foundation of an earlier tool called NIRVANA, Illumina Connected Annotations is part of the Illumina Connected Software suite. This cutting-edge genomic annotation engine combines the speed and accuracy of existing Illumina software solutions by directly integrating as a core component within key Illumina analysis and interpretation offerings (such as DRAGEN4 and Emedgene) while enabling the flexibility and customization necessary for breakthrough discovery.
What does Illumina Connected Annotations provide?
The annotations software reads in single or multi-sample VCFs for small variants (including MNVs), CNVs, SVs, STRs, and/or fusions, such as those generated by the DRAGEN DNA and RNA analysis pipelines.
Illumina Connected Annotations provides the predicted impact on coding sequence (“c.” or “cNomen”) and protein sequence (“p.” or “pNomen”) following Human Genome Variation Society (HGVS) standards,5 as well as consequences relevant to each variant using Sequence Ontology standard nomenclature.6 The transcript and gene models are obtained from RefSeq (version GCF_000001405.40-RS_2023_03, 2023-03-21) and Ensembl (release 110, 2023-04-27).
How does functional annotation work?
Functional annotation as part of Illumina Connected Annotations
- identifies transcripts intersecting each alternate allele using an interval array,
- marks overlapping exons and/or introns,
- adjusts for discrepancies between transcript and genomic reference sequences, and
- provides the cNomen and pNomen.
Canonical transcripts are identified using information from MANE7 or, when not available, via existing heuristic methods.8,9 This phase also provides consequences relevant to each variant using Sequence Ontology standard nomenclature. Illumina Connected Annotations performs right-alignment to coding and protein sequences as needed, in accordance with HGVS standards.5 If applicable, it also adds associated Cancer Hotspots annotation. Then it evaluates SVs to identify potential unidirectional gene fusions based on the resulting gene orientation and proximity. Known fusions are annotated using paired gene symbols from resources such as COSMIC10 and FusionCatcher.11 In the final phase, it adds gene-specific annotations for each unique gene with at least one variant in the VCF, retrieving data from OMIM, ClinGen, and other gene information sources.
Annotation Sources
Illumina Connected Annotations also integrates the most commonly used and powerful public databases, like gnomAD and COSMIC, as well as predictive scores developed at Illumina, like SpliceAI12and PrimateAI-3D.13 Read more about the power of genomic AI for breakthrough discovery, and how Illumina is helping to lead the way, in this article in Genetic Engineering & Biotechnology News.
The data sources supported are listed in Table 1 and divided into two tiers: Premium and Basic. The Basic tier can be accessed free of charge. The Premium tier requires a license. For access, please contact annotation_support@illumina.com.
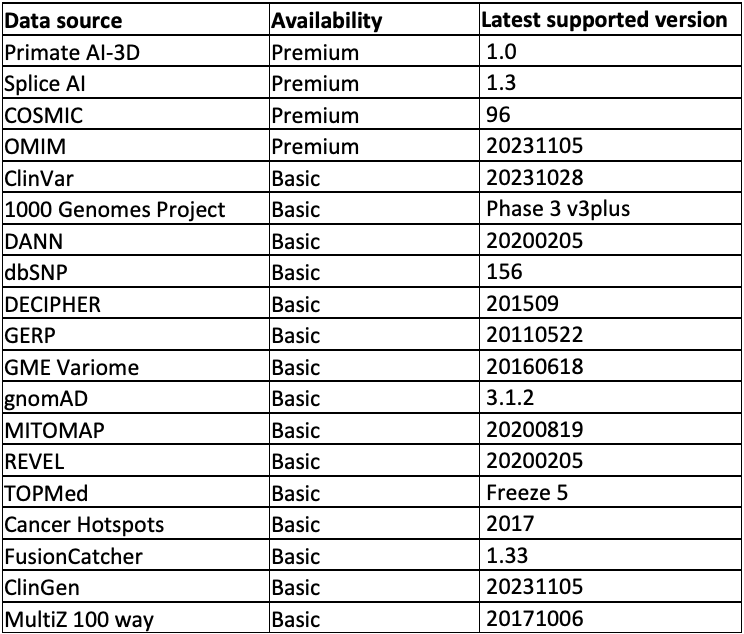
Table 1: Annotation sources supported out of the box from the Illumina Connected Annotations tool—Note that in addition to the data sources listed above, most annotation resources, including your own data, can be formatted into custom annotation resources. This table is kept up to date here.
Accuracy and speed
To measure accuracy, we compared the output of Illumina Connected Annotations to the HGVS results for a set of nearly 9 million variants called from whole-genome sequencing (WGS) and whole-exome sequencing (WES). We also compared the accuracy against third-party tools VEP,14 SnpEff,15 and ANNOVAR.16
The experiment entailed a direct comparison of the HGVS notations generated by each annotation tool against the HGVS notations provided by BioCommons, which is recognized as the gold standard.
Figure 1 shows the accuracy results of Illumina Connected Annotations, VEP, SnpEff, and ANNOVAR for HGVS genomic, coding, and protein notation for a WGS VCF (about 6.5 million variants).
- HGVS genomic accuracy: Illumina Connected Annotations correctly annotated all variants, achieving an accuracy of 100%, while VEP shows an accuracy of 99.970%
- HGVS coding accuracy: Illumina Connected Annotations shows an accuracy of 99.997%, while other tools range from 99.962 to 99.991%.
- HGVS protein accuracy: Illumina Connected Annotations presents a high accuracy rate of 99.998%, matching VEP’s accuracy and slightly outperforming SnpEff and ANNOVAR, which have accuracy rates of > 99.988%.
Overall, Illumina Connected Annotations exhibits high accuracy rates across all categories for HGVS notations.
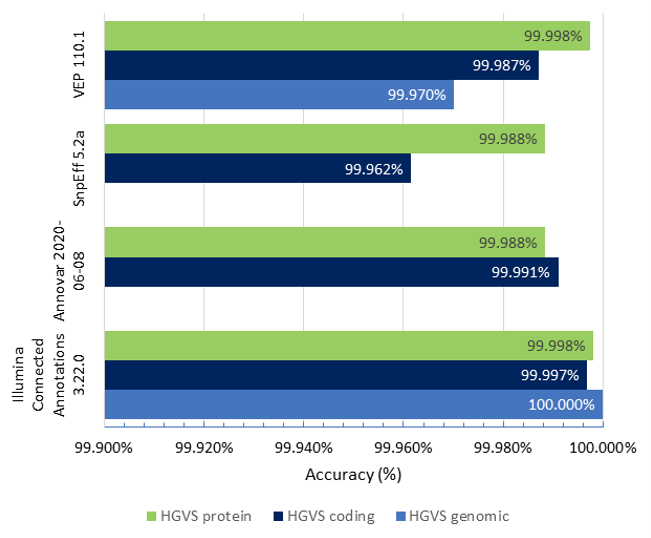
Figure 1: Accuracy of Illumina Connected Annotations, VEP, SnpEff, and ANNOVAR for HGVS genomic, coding, and protein notation for a WGS VCF, using the HGVS notations provided by BioCommons as the reference.
Illumina Connected Annotations has been heavily optimized for whole-genome analysis on a population scale, both for germline genetics and somatic oncology applications. Because a typical human genome has more than 4 million variants and tumor genomes can have far more than that, annotation speed can be a major bottleneck.
To show how Illumina Connected Annotations performs, we ran annotations for a whole-genome germline VCF that contains about 6.5 million variants. We also annotated the same VCF file using VEP, SnpEff, and ANNOVAR as a comparison. Figure 2 below shows the run time for each software to annotate about 6.5 million variants.
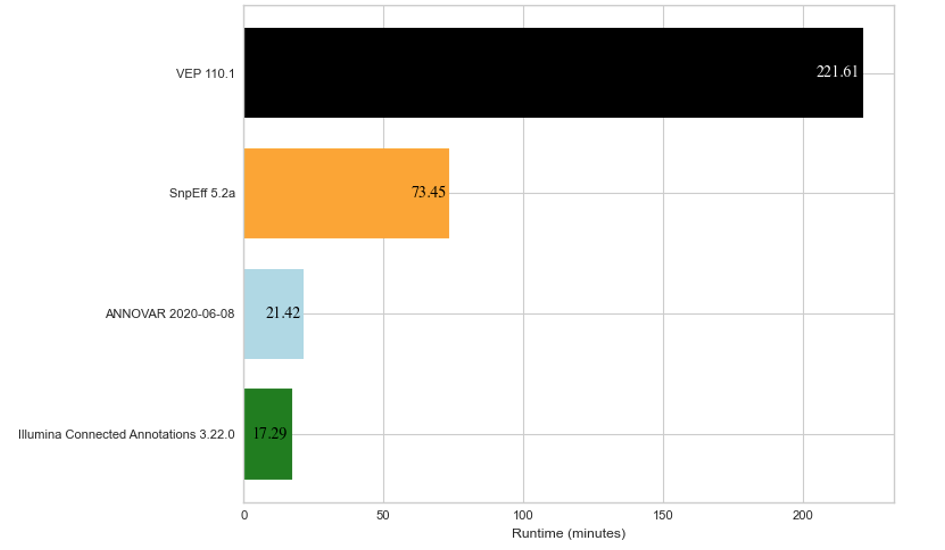
Figure 2: Average run time results for annotation of about 6.5 million DNA variants across three replicates for four different annotation algorithms.
The annotations compared above were each run on the same cloud-based hardware (AWS EC2 c5.4xlarge with 16vCPU and 32 GB of RAM). Note that SnpEff only annotates using the RefSeq database, while VEP, ANNOVAR, and Illumina Connected Annotations use both RefSeq and Ensembl databases.
Scaling to population genomics studies
Illumina Connected Annotations can annotate not only single sample VCFs but also multi-sample VCFs, as generated by DRAGEN Iterative gVCF Genotyper, for annotation at scale. To avoid copying large amounts of sample-level information into the annotation output JSON file, users may optionally create a site-level VCF containing only essential information extracted from the multi-sample VCF—namely CHROM, POS, REF, and ALT (ALT may contain multiple alleles). This site-level VCF can then be used as input to Illumina Connected Annotations, such that in the output file, the variants called across all the cohort samples are annotated once. Users may further split the site-level VCF by genomic regions to maximize the parallelization of the annotation process. We used this method to annotate 128 million variants called in The 1000 Genomes Project (3202 samples) and 1.5 billion variants called in the UK Biobank 500,000 whole genomes release (490,541 samples).2,3 Illumina Connected Annotations was also used to annotate the joint call set of more than 1 billion genetic variants in the All of Us Research Program.1
How to run
As displayed in Table 2 below, Illumina Connected Annotations can be run under various use cases—either as a standalone executable, or within DRAGEN Secondary Analysis. This includes cloud-based execution using Illumina Connected Analytics and BaseSpace Sequence Hub, and on-premise servers. There is no charge for the use of Premium-tier data sources within Illumina Connected Annotations for DRAGEN users.

Table 2: Deployment models of Illumina Connected Annotations—*To obtain a license for the Premium database, email annotation_support@illumina.com. †Download the standalone build here. ‡Available in DRAGEN version 4.3.
Join us in the genomic revolution
We invite the genomics community to explore Illumina Connected Annotations and its capabilities with the basic and premium data sources. Join us in pushing the boundaries of what's possible. Illumina Connected Annotations is more than a tool; it's a catalyst for progress, and we can't wait to see the transformative impact it will have on your genomic research endeavors.
Please get in touch with annotation_support@illumina.com if you want to learn more, use the tool, or have suggestions for additional resources we should incorporate into future versions. The standalone version of the tool is available for download here, with documentation available here.
References
- The All of Us Research Program Genomics Investigators. Genomic data in the All of Us Research Program. Nature. February 19, 2024. doi:10.1038/s41586-023-06957-x
- Halldorsson BV, Eggertsson HP, Moore KHS, et al. The sequences of 150,119 genomes in the UK Biobank. Nature. July 2022;607(7920):732-740. doi:10.1038/s41586-022-04965-x
- Science. UK Biobank releases half a million whole-genome sequences for biomedical research. science.org/content/article/uk-biobank-releases-half-million-whole-genome-sequences-biomedical-research. November 29, 2023. Accessed February 2024.
- Behera S, Catreux S, Rossi M, et al. Comprehensive and accurate genome analysis at scale using DRAGEN accelerated algorithms. bioRxiv. January 6, 2024. doi:10.1101/2024.01.02.573821
- den Dunnen JT. Describing Sequence Variants Using HGVS Nomenclature. Methods Mol Biol. 2017;1492:243-251. doi:10.1007/978-1-4939-6442-0_17
- Eilbeck K, Lewis SE, Mungall CJ, et al. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6(5):R44. doi:10.1186/gb-2005-6-5-r44
- Chaisson MJP, Sanders AD, Zhao X, et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun. April 16, 2019;10(1):1784. doi:10.1038/s41467-018-08148-z
- Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. January 4, 2016;44(D1):D862-D868. doi:10.1093/nar/gkv1222
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. May 2015;17(5):405-424. doi:10.1038/gim.2015.30
- Bamford S, Dawson E, Forbes S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. July 19, 2004;91(2):355-358. doi:10.1038/sj.bjc.6601894
- Beccuti M, Carrara M, Cordero F, et al. Chimera: a Bioconductor package for secondary analysis of fusion products. Bioinformatics. December 15, 2014;30(24):3556-3557. doi:10.1093/bioinformatics/btu662
- Jaganathan K, Panagiotopoulou SK, McRae JF, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell. January 24, 2019;176(3):535-548 e24. doi:10.1016/j.cell.2018.12.015
- Gao H, Hamp T, Ede J, et al. The landscape of tolerated genetic variation in humans and primates. Science. June 2, 2023;380(6648):eabn8153. doi:10.1126/science.abn8197
- McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. June 6, 2016;17(1):122. doi:10.1186/s13059-016-0974-4
- Cingolani P. Variant Annotation and Functional Prediction: SnpEff. Methods Mol Biol. 2022;2493:289-314. doi:10.1007/978-1-0716-2293-3_19
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. September 2010;38(16):e164. doi:10.1093/nar/gkq603